











SWISS-MODEL
SWISS-MODEL,自动化蛋白质结构同源建模服务器,面向全球生命科学研究人员提供蛋白质三维结构模型构建服务
标签:数据可视 生信分析SWISS-MODEL SWISS-MODEL官网 SWISS-MODEL官网入口SWISS-MODEL官网,全自动蛋白质结构同源建模服务器,面向全球生命科学研究者
什么是SWISS-MODEL?
SWISS-MODEL是一个全自动化的蛋白质结构同源建模服务器,由瑞士生物信息学研究所与巴塞尔大学联合开发,自1993年开创该领域以来,已成为全球应用最广泛的免费在线蛋白质三维结构预测平台。该平台的核心功能是基于同源建模原理,利用已知实验解析的蛋白质结构作为模板,通过BLAST和HHblits等工具在源自蛋白质数据库的模板库中搜索最佳匹配,然后执行序列比对、刚体片段组装与能量优化,自动构建目标蛋白的精确三维模型。其特色在于提供了三种不同控制级别的建模模式:全自动模式仅需提交氨基酸序列或UniProt编号即可一键生成模型;对齐模式允许用户上传自定义的序列比对结果;项目模式则结合DeepView可视化工具,便于专家对复杂案例进行手动校正。完成建模后,系统会通过QMEAN统计势能函数提供全局与局部模型质量评分、拉氏图评估及寡聚状态预测,帮助用户判断模型的可靠性。此外,SWISS-MODEL还包含一个持续更新的模型仓库,基于最新UniProtKB数据为人类、小鼠、大肠杆菌、新冠病毒等十几种模式生物及病原体的蛋白质组提供预计算的高质量三维模型,并与UniProt、STRING等主流数据库深度整合。该平台每年处理超过两百万次建模请求,并已整合AlphaFold数据库结构作为补充模板,使其预测能力更加强大,广泛服务于全球生命科学领域的研究人员。
SWISS-MODEL官网: https://swissmodel.expasy.org/
SWISS-MODEL深度评测:2026年,生物学家手中的蛋白质结构预测利器
一、 引言
在生命科学的研究中,蛋白质是执行生命活动的主力分子。理解一个蛋白质的功能,最直接的方式就是看清它的三维结构。然而,通过 X 射线晶体学、冷冻电镜或核磁共振等实验手段解析蛋白质结构,至今仍是一项耗时、昂贵且成功率不确定的工作。目前,UniProt 数据库中已知的蛋白质序列数量已超过 2 亿条,而已被实验解析的蛋白质结构数量却仅有 20 余万,两者之间横亘着一条巨大的“序列-结构鸿沟”。
这正是计算结构生物学大显身手的舞台。在众多计算工具中,同源建模(Homology Modeling,又称比较建模)是填补这条鸿沟最准确、最可靠的方法。它的核心逻辑朴素而有力:如果两个蛋白质的氨基酸序列相似,那么它们很可能拥有相似的三维结构。基于这一原理,只要找到一个已知结构的“模板”,就可以为未知结构的“目标”蛋白质搭建出可信的三维模型。
而在这个领域,有一个名字是无法绕过的常青树——SWISS-MODEL。作为世界上第一个全自动蛋白质同源建模服务器,它自 1993 年诞生以来,已经走过了三十多年的历程。它不仅是结构生物信息学领域被引用次数最多的工具之一(截至 2015 年就已被引用超过 20000 次),更在 2026 年的今天,依然保持着每年处理超过 200 万次建模请求的惊人活力,持续为数以万计的生命科学研究者提供着关键的结构信息。
进入 2026 年,蛋白质结构预测的版图已因 AlphaFold 等 AI 工具的出现而发生了翻天覆地的变化。那么,SWISS-MODEL 这个经典的“老将”是否已经过时?它在与 AI 预测工具的竞争中,找到了怎样的新定位?它的建模质量是否依然可靠?其操作流程对于非生物信息学专业的“湿实验”研究者是否足够友好?
本文将为你带来一份全面、深度、客观的 SWISS-MODEL 使用指南与测评报告。我们将从其底层原理和最新动态出发,深度拆解其三大核心工作模式,手把手教你如何操作,并对其性能、体验进行真实评测。同时,我们还会将其与 I-TASSER、AlphaFold、Modeller 等主流竞品进行多维度横向对比,分析价格方案,并给出明确的选购与使用决策建议。无论你是刚接触结构生物学的学生,还是希望快速获取蛋白质结构信息以指导实验的资深研究者,这篇文章都将是你的得力助手。

二、 什么是SWISS-MODEL
SWISS-MODEL 是一款全自动的、基于网络的蛋白质结构同源建模专家系统。它由瑞士生物信息学研究所(SIB)和巴塞尔大学 Biozentrum 中心的 Torsten Schwede 教授课题组开发并维护,通过 ExPASy 门户网站向全球免费开放。其核心使命简单而明确:让任何生命科学研究者,无论是否具备计算结构生物学的专业知识,都能轻松、快速地根据蛋白质的氨基酸序列,生成可靠的三维结构模型。
你不需要安装任何软件,也不需要配置复杂的计算环境。只需打开浏览器,输入目标蛋白质的氨基酸序列或 UniProt 数据库编号,SWISS-MODEL 就会在后台自动执行一整套严谨的建模流程:它会在一个持续更新的模板库(SMTL)中搜索合适的已知结构作为模板,进行序列比对,然后通过刚体片段组装技术搭建模型,最后利用 QMEAN 等统计势能函数对模型质量进行量化评估。整个过程从提交到获得结果,通常在几分钟到几十分钟内即可完成。
SWISS-MODEL 不仅仅是一个建模工具,它更是一个由三个紧密集成的组件构成的生态系统:
- SWISS-MODEL 管线:一套包含模板识别、比对、建模和质量评估的全自动软件工具与数据库。
- SWISS-MODEL 工作区:一个基于网页的图形化用户界面,为用户提供了从简单到复杂的多种交互模式。
- SWISS-MODEL 仓库:一个持续更新的数据库,为人类、小鼠、大肠杆菌、新冠病毒等高生物医学价值的模式生物,预先计算并存储了海量的蛋白质同源模型。
简单来说,SWISS-MODEL 就像一位不知疲倦的、拥有三十年经验的资深结构生物学家,全天候待命,随时准备为你将一维的序列信息,翻译成直观、可交互的三维结构模型。
三、 目标客户和应用场景
SWISS-MODEL 的设计哲学是“服务于所有生命科学研究者”,但这并不意味着它对所有人都同样适用。精准地理解其目标用户画像和典型的应用场景,能帮助你判断它是否是你工具箱中必备的那把“螺丝刀”。
1. 核心目标客户画像
SWISS-MODEL 最核心的用户群体,是那些需要蛋白质结构信息来驱动和解释实验,但本身并非计算结构生物学专家的“湿实验”研究者。他们可能是分子生物学家、生物化学家、药理学家或药物化学家。对于他们而言,SWISS-MODEL 的价值在于提供了一个极低门槛的入口,能够快速地将结构洞察转化为可验证的实验假设。
| 目标客户群体 | 行业/岗位 | 核心需求 | 推荐指数 |
|---|---|---|---|
| 分子生物学家 | 学术机构、生物技术公司 | 设计点突变实验,理解突变如何影响蛋白功能与稳定性;预测蛋白结构域边界。 | ★★★★★ |
| 生物化学家 | 高校、研究所、制药企业 | 理解酶催化机制;分析蛋白-配体相互作用;辅助蛋白纯化和结晶实验设计。 | ★★★★★ |
| 药理学家/药物化学家 | 制药公司、CRO | 基于结构的药物设计(SBDD)初筛;分析靶点蛋白的可成药口袋;指导先导化合物优化。 | ★★★★☆ |
| 计算生物学家(入门级) | 学术界、生物信息公司 | 作为复杂计算管线的起点;快速获取模型用于后续分子动力学模拟或对接初筛。 | ★★★★☆ |
| 教育工作者与学生 | 大学、研究所 | 教学演示蛋白质结构与功能的关系;在课程中实践生物信息学分析。 | ★★★★★ |
| 进化生物学家 | 学术机构 | 比较近缘或远缘物种间蛋白结构差异,推测功能演化。 | ★★★☆☆ |
2. 典型应用场景一:指导关键氨基酸残基的功能验证实验
场景描述:你正在研究一个与肿瘤发生相关的未知功能蛋白 X。通过序列分析,你怀疑它可能是一个激酶,但具体的催化位点和底物结合位点尚不明确。你计划通过丙氨酸扫描突变来逐一验证关键残基的功能,但盲目突变整个蛋白耗时耗力。
使用方式与效果:你只需将蛋白 X 的序列提交到 SWISS-MODEL。系统在几分钟内生成了一个高质量的模型,模板是一个已知的激酶结构。通过观察模型,你清晰地识别出了 ATP 结合口袋中的几个保守残基(如催化 loop 中的 Asp,活化 loop 中的磷酸化位点 Ser/Thr),以及一个可能负责底物识别的带正电的裂隙。你立刻将突变实验聚焦在这 10-15 个候选残基上,而不是全蛋白的 300 多个残基,实验效率提升了数十倍。随后,你将关键残基的突变体进行体外活性测试,结果完美验证了你的模型预测。
3. 典型应用场景二:分析疾病相关突变的结构基础
场景描述:临床合作者发现了一个与罕见遗传病相关的错义突变(例如,将某个蛋白第 237 位的甘氨酸突变为精氨酸,G237R)。你需要从分子层面解释,为什么这个小小的改变会导致如此严重的功能丧失。
使用方式与效果:你获取野生型蛋白序列,通过 SWISS-MODEL 的“自动模式”快速获得其三维模型。然后,你利用 SWISS-MODEL Workspace 的“项目模式”,或者将模型下载后在 DeepView(Swiss-PdbViewer)中打开,手动将第 237 位的甘氨酸突变为精氨酸。结构分析一目了然:甘氨酸位于一个紧密的 β-转角处,其独特的柔性和小侧链是维持该转角构象的关键。而庞大的、带正电的精氨酸侧链在空间上完全无法被容纳,会与周围疏水核心发生剧烈冲突,导致整个蛋白结构域的错误折叠和降解。这个直观的结构解释,为后续的体外表达和热稳定性实验提供了完美的分子假说。
4. 典型应用场景三:为分子对接和虚拟筛选准备靶点结构
场景描述:你的团队发现了一个极具潜力的新型抗菌靶点蛋白 Y,但尚未解析其晶体结构。你想在等待晶体学结果的同时,快速启动计算机辅助药物发现,利用商业化合物库进行虚拟筛选,寻找先导化合物。
使用方式与效果:你使用 SWISS-MODEL 为蛋白 Y 构建了高质量的同源模型。模型的质量评估分数(QMEAN)很好,并且与同家族蛋白的活性位点高度保守。你将此模型作为受体,使用 SwissDock(同样来自 SIB 的免费对接工具)或商业软件,对一个包含数百万化合物的库进行了大规模虚拟筛选。虽然同源模型在侧链构象精度上无法与晶体结构媲美,但足以用于过滤掉明显不匹配的化合物,将筛选范围缩小到几百个候选分子。这为后续的购买、体外活性测试赢得了宝贵的数月时间,极大地加速了药物发现的早期进程。
5. 不适合哪些人?
尽管功能强大,SWISS-MODEL 并非万能钥匙,以下用户群体可能会觉得它“不够用”或“不适用”:
- 需要极高原子精度模型的研究者:如果你的目标是研究精细的催化机理、进行精确的自由能微扰计算,或者需要分辨率优于 1.5 Å 的侧链细节,那么同源模型,尤其是基于低序列一致性模板的模型,其固有的误差是无法满足需求的。此时,X 射线晶体学或冷冻电镜实验才是最终解决方案。
- 对全新折叠(De Novo Folding)蛋白的研究者:SWISS-MODEL 的核心是同源建模,它必须依赖一个已知结构的模板。如果你的目标蛋白序列在 PDB 数据库中找不到任何同源结构(即所谓的“孤儿蛋白”),SWISS-MODEL 将无法提供有效模型。此时,你需要求助于 AlphaFold2/3、RoseTTAFold 等基于深度学习的从头预测工具。
- 需要深度定制和批量化建模的高级计算生物学家:虽然 SWISS-MODEL 提供了 API 和项目模式,但对于需要高度自定义建模流程(如使用非标准力场、复杂的多模板建模、大规模跨膜蛋白建模)的专业用户,本地化的 Modeller 或 Rosetta 软件套件提供了无与伦比的灵活性和控制力。
| 应用场景 | 适配工具 | 使用方式 | 预期效果 | 难度等级 |
|---|---|---|---|---|
| 快速获得蛋白结构概览 | SWISS-MODEL | 提交序列,全自动建模 | 几分钟内获得直观的 3D 模型和初步质量评估 | ★☆☆☆☆ |
| 指导点突变功能实验 | SWISS-MODEL | 建模后,在 DeepView 中分析突变位点 | 准确定位功能残基,大幅减少实验试错成本 | ★★☆☆☆ |
| 解释疾病突变的结构后果 | SWISS-MODEL | 建模后,手动引入突变并分析构象张力 | 提供强有力的分子机制假说,指导后续实验 | ★★☆☆☆ |
| 准备虚拟筛选的受体结构 | SWISS-MODEL | 建模后,准备 PDB 文件用于对接软件 | 快速启动 SBDD 流程,缩小候选化合物范围 | ★★★☆☆ |
| 预测全新折叠蛋白结构 | AlphaFold2/3 | 使用在线服务或本地安装 | 高精度地预测无模板蛋白的全新结构 | ★★★★☆ |
| 大规模、高度定制的批量化建模 | Modeller | 编写 Python 脚本,本地运行 | 实现复杂、多模板、含配体的全自定义建模管线 | ★★★★★ |
四、 核心功能深度拆解
SWISS-MODEL 的强大之处,在于它将一个复杂的、多步骤的专家级任务,封装成了三种不同交互深度的模式,让用户可以根据自己的需求和专业水平自由选择。本章节将对这些核心功能进行“手把手教学+深度评测”式的拆解。
1. 杀手级功能一:自动模式——一键成模的极致便捷
这是 SWISS-MODEL 的门面,也是绝大多数用户(超过 90%)使用的模式。它的设计哲学是**“你只需提供序列,剩下的交给我们”**。
完整功能介绍:在自动模式下,用户仅需提供目标蛋白的氨基酸序列(FASTA 格式或纯文本)或其 UniProt 数据库编号。提交任务后,服务端的管线将全自动执行以下四步严谨流程:
- 模板搜寻:系统会使用 BLAST 和 HHblits 两种算法,在 SWISS-MODEL 模板库(SMTL)中搜索同源模板。SMTL 是一个从 PDB 数据库精心整理并定期更新的高质量模板库,确保了模板的质量和可追溯性。
- 序列比对:选定最佳模板后,系统会自动生成目标序列与模板结构的比对。
- 模型搭建:SWISS-MODEL 采用“刚体片段组装”方法。它将目标序列拆解为小的片段,在模板结构的约束下,通过组装这些片段来构建模型,并进行能量最小化以消除不合理的原子冲突。
- 质量评估:模型搭建完成后,系统会使用内置的 QMEAN 综合评分系统对模型进行全局和局部质量评估,并以直观的图表形式呈现。
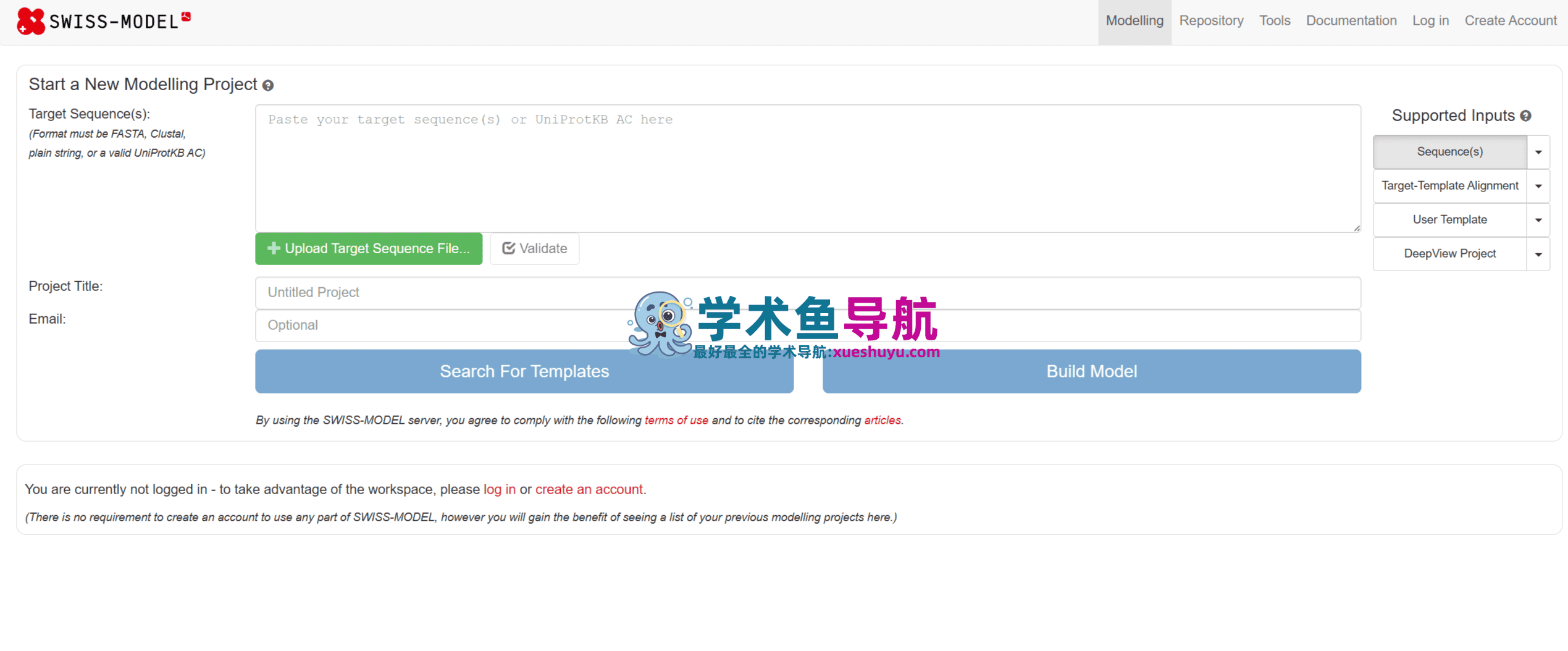
操作步骤:
- 访问 SWISS-MODEL 官网(swissmodel.expasy.org),点击“Start Modelling”。
- 在弹出的窗口中,将你的目标蛋白序列粘贴到输入框,或者输入 UniProt AC(如 P00533),然后点击“Build Model”。
- 等待几分钟到半小时(取决于蛋白大小和服务器负载)。你可以在“My Jobs”页面查看任务进度。
- 任务完成后,你将看到一个结果页面,包含:
- 模型三维视图:一个交互式的 3D 分子可视化窗口。
- 建模报告:详细列出了所用的模板、序列一致性、覆盖范围等信息。
- 质量评估图:一个彩色的条带图,直观显示模型每个残基的预期质量(QMEAN 局部打分),以及全局质量分数与参考结构的比较。
- 下载选项:可下载模型的 PDB 文件、建模报告等。
使用技巧与最佳实践:
- 优先使用 UniProt ID:如果你研究的是 UniProt 数据库中的标准蛋白,直接输入 ID 是最佳选择,因为系统会自动获取其规范的序列和异构体信息,避免手动输入错误。
- 理解模板质量:建模结果的好坏 80% 取决于模板。务必关注“Seq Identity”(序列一致性)和“Coverage”(覆盖度)。序列一致性 > 50% 的模型通常非常可靠,可以用于药物设计等精细应用;30%-50% 的模型整体折叠通常是正确的,但 loop 区等细节误差较大;< 30% 的模型则需要非常谨慎地解读。
- QMEAN 分数的正确解读:QMEAN 分数并非绝对的“好”与“坏”,而是告诉你模型与实验结构的相似程度。-4.0 以下是一个警示信号,但一个 -2.0 的分数可能意味着你的蛋白本身就是一个高度柔性的、部分无序的蛋白,而不是模型质量差。务必结合 QMEAN 的局部打分图来观察哪些区域不可靠。
与同类功能的对比:
| 功能对比 | SWISS-MODEL 自动模式 | I-TASSER 在线版 | AlphaFold2 (ColabFold) |
|---|---|---|---|
| 操作门槛 | 极低,粘贴序列即可 | 较低,需填写表单 | 中等,需运行 Notebook |
| 建模速度 | 快(分钟级) | 慢(小时级到天级) | 中等(分钟到小时级) |
| 核心算法 | 同源建模(模板依赖) | 穿线法+从头模拟 | 深度学习(无需模板) |
| 结果呈现 | 直观的网页报告,质量评估清晰 | 网页报告,多种评分 | PDB 文件 + PAE 图,需自备可视化工具 |
| 适用场景 | 有同源模板的快速建模 | 难建模的远缘同源目标 | 通用、高精度,尤其适合无模板目标 |
| 稳定性与可靠性 | 极高,30年验证 | 高,但服务器队列长 | 依赖 Google Colab 资源,偶有波动 |
2. 杀手级功能二:比对模式——掌控权的适度下放
当自动模式选择的模板或比对结果不尽如人意时,比对模式(Alignment Mode)为你提供了一个优雅的中间地带。它允许你输入自己生成的序列比对结果,从而跳过模板搜索和自动比对步骤,直接进入模型搭建。
完整功能介绍:在此模式下,用户负责提供目标序列与模板序列的比对(FASTA 格式)。你可以使用任何你信赖的比对工具(如 Clustal Omega, T-Coffee, MUSCLE)来完成这一步,并根据自己的生物学知识进行手动调整。SWISS-MODEL 会严格基于你提供的比对来搭建模型。
操作步骤:
- 首先,你需要自行完成目标序列与一个或多个模板的比对,并准备好包含目标序列和模板序列的 FASTA 格式比对文件。
- 在 SWISS-MODEL 首页选择“Alignment Mode”。
- 将你的比对粘贴到输入框,并指定哪条是目标序列,哪条是模板。
- 提交任务。系统将基于此比对进行模型搭建和质量评估。
真实使用感受与效率提升:这是一个非常“务实”的功能。例如,在研究一个蛋白家族时,你清楚地知道某个特定的 loop 区在家族成员中长度和构象差异很大,自动比对可能会在此区域产生错误。通过使用比对模式,你可以手动调整该区域的比对,引入适当的 gap,确保关键功能区域的模型质量。这避免了完全手动建模的繁琐,又将关键的生物学洞察注入到了自动化流程中,效率提升显著。你将模型从“可能对”变成了“大概率对”。
最佳实践:
- 模板选择的艺术:在选择模板时,不要只看序列一致性。要考虑模板的构象状态(是激活态还是失活态?)、分辨率、是否含有配体、以及覆盖范围。有时,一个序列一致性稍低但构象状态更相关的模板,是更好的选择。
- 多模板建模的局限:SWISS-MODEL 的比对模式主要基于单模板。虽然其项目模式支持多模板,但在比对模式下,你提供的是一个单一的比对,因此模型主要基于该比对中的模板搭建。对于需要多模板建模的复杂情况,项目模式或 Modeller 是更好的选择。
3. 杀手级功能三:项目模式——解锁专业级控制
项目模式(Project Mode)是 SWISS-MODEL 为高级用户准备的终极武器。它通过与其桌面端可视化工具 DeepView (Swiss-PdbViewer) 的深度集成,将手动干预的能力提升到了极致。
完整功能介绍:项目模式的核心是一个 DeepView 项目文件。DeepView 是一款功能强大的免费蛋白质结构可视化和分析软件。在项目模式中,你的工作流程变为:
- 在 DeepView 中加载模板结构和目标序列。
- 使用 DeepView 的“魔术拟合”工具和手动调整功能,在结构环境中对目标-模板比对进行迭代式优化。
- 保存为一个包含了所有修改信息的项目文件。
- 将此项目文件提交到 SWISS-MODEL Workspace。
操作步骤:
- 下载并安装 DeepView。
- 从 PDB 下载模板文件并在 DeepView 中打开。
- 通过菜单
SwissModel->Load Raw Sequence to Model加载目标序列。 - 使用
Fit->Magic Fit进行初步结构比对,然后手动调整比对中的错配和 Gap 位置。你可以实时看到调整比对对模型结构产生的潜在影响。 - 调整满意后,通过
SwissModel->Submit Modeling Request直接提交项目文件。 - 模型完成后,结果会发送到你预留的邮箱,并可在 Workspace 中查看。
常见误区:
- 误区:项目模式就是手动建模。 实际上,项目模式的核心仍然是手动优化比对,而非手动搭建模型。模型的搭建和能量最小化依然由 SWISS-MODEL 自动完成。它解决的是“输入”问题,而不是“算法”问题。
- 误区:任何情况都需要用项目模式。 对于序列一致性 > 50% 的简单任务,自动模式已经足够好,手动干预带来的提升微乎其微,反而会浪费时间。项目模式的价值在于处理那些 30%-50% 序列一致性的“灰色地带”任务。
4. 差异化特色功能:同源寡聚体与关键配体的自动化建模
这是 SWISS-MODEL 区别于许多其他简易建模工具的核心亮点,也是它保持前沿性的关键。蛋白质在体内很少以单体形式工作,它们常常形成二聚体、四聚体等复杂的四级结构,其功能也与金属离子、辅因子等配体密切相关。
功能详解:SWISS-MODEL 的自动模式能够“理解”并重现模板中的四级结构和关键配体。
- 同源寡聚体建模:如果模板是一个同源二聚体,SWISS-MODEL 会自动为你的目标序列搭建一个二聚体模型,而不是简单的单体。它会精确地模拟亚基间的相互作用界面。这对于研究信号通路中受体的二聚化激活、酶的别构调控等至关重要。
- 配体建模:模板中结合的关键金属离子(如 Zn²⁺, Mg²⁺)或生物相关配体(如 ATP, NAD⁺, Heme),会被自动转移到目标模型中。这不仅仅是简单的复制坐标,而是会进行局部能量优化,确保配体与目标蛋白残基之间的几何构型合理。
为什么这个功能让它脱颖而出? 许多其他在线建模工具只输出一个“裸”的单体蛋白模型。而 SWISS-MODEL 提供的模型直接包含了功能相关的四级结构和配体信息,这使得模型在生物学上更具“可用性”。例如,你构建了一个蛋白激酶的模型,它直接就是一个带有 ATP 和两个 Mg²⁺ 的活性构象二聚体。你可以立即将其用于对接或突变分析,而无需再手动查找和添加辅因子,也无需猜测其活性寡聚状态。这极大地提升了模型的下游应用价值,体现了 SWISS-MODEL 作为一个“专家系统”的深厚积淀。
5. 针对高级用户的隐藏技巧
抛开图形界面,SWISS-MODEL 为追求自动化和集成的用户提供了强大的编程接口。
- REST API:SWISS-MODEL 提供了 RESTful API,允许你通过编程方式提交建模任务、查询任务状态和下载结果。你可以将 SWISS-MODEL 无缝集成到你自己的生物信息学管线中,实现批量化、自动化的建模。例如,你可以编写一个 Python 脚本,读取一个包含 100 个蛋白序列的文件,循环提交任务,并自动收集所有模型的 QMEAN 分数进行统计分析。
- OpenStructure Actions:根据 2026 年的最新动态,SWISS-MODEL 背后的核心计算引擎 OpenStructure 的功能,已经可以通过 API 以“Actions”的形式进行访问。这意味着你可以直接在云端调用 SWISS-MODEL 所使用的模板搜索、比对、模型搭建等原子化功能模块,构建高度定制化的建模流程,而无需在本地编译复杂的 OpenStructure 源码。这为高级用户提供了前所未有的灵活性和计算能力。
- SWISS-MODEL Repository 的直接利用:不要只把 Repository 当作一个浏览数据库。你可以通过其物种分类页面,直接下载某一物种(如人类)所有预先计算好的模型文件。这对于需要大规模结构数据的分析(如蛋白质组范围的结构功能注释、系统生物学建模)来说,是一个巨大的宝藏,省去了你逐条建模的繁琐过程。
6. 功能完整度评估
| 核心功能 | 支持情况 | 备注与替代方案 |
|---|---|---|
| 单模板自动建模 | ✅ 完美支持 | 核心功能,体验极佳。 |
| 用户自定义比对建模 | ✅ 完美支持 | 比对模式,提供中等控制权。 |
| 基于 DeepView 的项目模式 | ✅ 完美支持 | 提供最高级别的手动控制,但需安装桌面软件。 |
| 同源寡聚体建模 | ✅ 自动支持 | 基于模板四级结构自动搭建,无需额外操作。 |
| 关键配体建模 | ✅ 自动支持 | 金属离子、辅因子等自动转移和优化。 |
| 模型质量评估 (QMEAN) | ✅ 完美支持 | 提供全局和局部打分,非常直观。 |
| 多模板建模 | ⚠️ 部分支持 | 在项目模式中可手动实现,但无自动多模板优化功能。替代方案:Modeller。 |
| 异源寡聚体建模 | ⚠️ 有限支持 | 需要更复杂的手动操作。替代方案:AlphaFold-Multimer。 |
| 非标准氨基酸/配体建模 | ❌ 受限 | 需要手动定义参数。替代方案:使用 SwissSidechain 数据库配合 Modeller。 |
| 大规模跨膜蛋白建模 | ⚠️ 一般 | 自动模式效果尚可,但精细调整困难。替代方案:RosettaMP。 |
| 蛋白质-核酸复合物建模 | ❌ 不支持 | 这是其明确边界。替代方案:HADDOCK, AlphaFold3。 |
| 从头结构预测(无模板) | ❌ 不支持 | 这是其原理所决定的。替代方案:AlphaFold2/3, RoseTTAFold。 |
五、 真实使用体验与深度测评
在本章节,我将从一个每周都会使用 SWISS-MODEL 的研究者视角,分享最真实的使用感受,不吹不黑。
1. 交互体验与UI设计
SWISS-MODEL 的网页界面完美诠释了“少即是多”的设计哲学。它没有花哨的动画,没有复杂的菜单层级,主页上硕大的“Start Modelling”按钮直指核心任务。这种极简主义风格对于其目标用户——那些只想快速得到结果而不想学习复杂软件的研究者——来说,是巨大的福音。
任务提交过程流畅自然,进度反馈清晰。结果页面是所有在线建模工具中,信息呈现最合理、最直观的之一。特别是QMEAN 质量评估条,用红-橙-蓝-绿的渐变色带,将每个残基的局部质量一目了然地映射到序列上,这个设计堪称经典。点击任何一个质量不佳的区域,3D 视图会立刻高亮显示该区域,让你能立刻聚焦到问题所在。
当然,缺点也在于此。对于习惯了 Ribbon 式或深色模式的现代 UI 的年轻用户来说,SWISS-MODEL 的界面可能显得有些“学院派”或“过时”。DeepView 的界面更是停留在 2000 年代初的风格,学习曲线陡峭,这是其项目模式普及的最大障碍。不过,考虑到其强大的功能完全免费,这种对功能而非颜值的偏执,是完全可以理解和接受的。
2. 性能与响应速度实测
在 2026 年的网络环境下,SWISS-MODEL 的服务器性能表现令人满意。对于绝大多数大小适中的蛋白(300-500 个氨基酸残基),在自动模式下,从提交到获得结果通常在 3-15 分钟 内完成。这相比于 I-TASSER 动辄数小时甚至数天的排队时间,优势是压倒性的。
这种速度优势主要源于其算法的高效。同源建模本质上比基于物理的模拟或深度学习推理要轻量得多。此外,SIB 维护的服务器基础设施显然也非常稳健,在我多年的使用经历中,极少遇到服务器宕机或任务卡死的情况。对于需要快速迭代假设的研究场景,这种及时反馈的价值是巨大的。
3. SWISS-MODEL优缺点对比
核心优势:
- 无与伦比的易用性:真正的“傻瓜式”操作,将极其复杂的流程简化到了极致,是连接“湿实验”与“干实验”的最佳桥梁。
- 极速响应:分钟级的建模速度,极大地加速了“序列→结构→假设→实验”的科研循环。
- 完全免费:对学术界永久免费,没有任何隐藏收费,极大地降低了结构生物学的入门门槛。
- 可靠性与稳定性:经过三十年的持续开发、维护和基准测试(CAMEO),其算法和服务器都极其成熟稳定,结果可信。
- 功能全面的“模型”而非“骨架”:自动包含寡聚体和关键配体,使模型具有直接的生物学功能意义,这是许多竞品不具备的。
- 卓越的质量评估体系:QMEAN 评分系统直观、易懂,能有效指导用户判断模型的可用范围,避免误用。
- 丰富的外部资源集成:与 UniProt, STRING, InterPro 等资源的深度链接,使得从一个蛋白出发进行多维度的系统分析变得非常便捷。
- 前瞻性的进化能力:积极整合 AlphaFold DB 作为模板,体现了其在新时代背景下,将 AI 预测与经典方法相结合的智慧,持续保持工具的先进性和生命力。
不足之处:
- 先天局限,无法逾越:作为同源建模工具,它无法预测全新折叠的蛋白结构。这是它的“能力边界”,而非“缺陷”。用户需要理解这一点,并在必要时转向 AlphaFold 等工具。
- 高级模式的现代化不足:DeepView 桌面软件的功能虽然强大,但其用户界面和交互逻辑已显陈旧,学习成本高,这阻碍了更多用户去探索项目模式的强大力量。我们期待一个基于网页的、现代化的 DeepView 替代品出现。
- 对非标准生物学的支持有限:在处理翻译后修饰、非天然氨基酸、或复杂的蛋白-核酸大复合物时,SWISS-MODEL 的能力捉襟见肘。这是其追求自动化、标准化的必然代价。
- 多模板建模的自动化程度有待提高:虽然手动可以实现,但缺少一个像 Modeller 那样智能的、自动化的多模板优化流程,可能会在某些复杂案例中错失最佳模型。
总的来说,这些不足之处更像是对一个优秀工具的“精益求精”的期许,而非影响其核心价值的致命伤。在它所设定的目标——快速、可靠、免费地为有模板的蛋白提供高质量同源模型——上,SWISS-MODEL 的表现依然是世界顶级的,是每一位生命科学研究者都值得信赖的伙伴。
六、 价格方案与性价比分析
对于学术用户而言,SWISS-MODEL 的性价比是无与伦比的,因为它是完全免费的。但对于商业用户,情况则有所不同。
1. 免费版 vs 付费版区别
SWISS-MODEL 的免费使用范围明确限定于学术界。其网站上明确注明了使用条款,要求商业用户联系开发团队获取授权。
| 项目 | 免费版(学术用途) | 商业授权版 |
|---|---|---|
| 适用对象 | 大学、研究所等非盈利性学术机构的师生和研究人员。 | 制药、生物技术、农业、化工等所有盈利性公司。 |
| 功能 | 访问全部功能:自动模式、比对模式、项目模式、API、Repository。 | 与免费版功能完全一致,无额外特权。 |
| 使用限制 | 无任务数量或计算资源上的硬性限制,但需遵守合理使用原则。 | 根据商业授权协议而定,通常会保证更高的服务级别协议(SLA)和专属支持。 |
| 技术支持 | 社区支持,通过帮助文档和邮件列表。响应速度不确定。 | 可获得来自 SIB 开发团队的优先、专业的技术支持。 |
| 价格 | 0 元 | 需联系 SIB 商业团队获取报价,通常根据公司规模和需求定制。 |
2. 哪个套餐最值得买?
对于学术用户,这个问题不存在,免费的午餐就在眼前,尽情享用即可。但请务必在发表成果时,正确引用 SWISS-MODEL 的相关文献,这是对其持续发展的最好支持。
对于商业用户,购买商业授权不仅是法律合规的要求,更是一笔明智的“投资”。你购买的不仅是软件使用权,更是:
- 法律风险规避:避免因未授权使用带来的知识产权和商业信誉风险。
- 可靠的支持保障:当你的药物研发管线高度依赖某个模型的准确性时,能够直接与开发团队沟通,获得专业解答,其商业价值远超授权费本身。
- 定制化可能:商业合作往往可以开启定制化开发或本地化部署的讨论,以满足公司特殊的数据安全或高通量需求。
3. 有无隐藏费用或退款政策?
SWISS-MODEL 没有任何隐藏费用,免费就是真免费。关于商业授权的具体商务条款、费用和退款政策,需要直接与 SIB 的技术转移办公室或其指定的商业代表进行沟通,官网上并无公开信息。
七、 竞品横向对比
在蛋白质结构预测的广阔天地里,SWISS-MODEL 并非孤军奋战。了解其主要竞品的优劣,能让你在合适的场景下做出最正确的选择。
1. I-TASSER vs SWISS-MODEL
I-TASSER 是另一个传奇级的在线蛋白质结构预测服务器,由密歇根大学张阳教授实验室开发。它曾在多次蛋白质结构预测关键评估(CASP)竞赛中名列前茅。
| 对比维度 | SWISS-MODEL | I-TASSER |
|---|---|---|
| 核心算法 | 同源建模(比较建模) | 穿线法(Threading)+ 从头模拟(Ab initio) |
| 模板依赖 | 强依赖,必须有可识别同源模板 | 弱依赖,即使无同源模板也能尝试预测 |
| 建模速度 | 快(分钟级) | 慢(小时到天级,服务器排队严重) |
| 易用性 | 极高,界面极简 | 中等,参数选项较多,新手易迷茫 |
| 模型质量(有模板时) | 高,尤其在高序列一致性下 | 高,与 SWISS-MODEL 相当 |
| 模型质量(无模板时) | 无法建模 | 可提供低到中等精度的模型 |
| 结果解读 | 非常直观,QMEAN 图一目了然 | 提供 C-score, TM-score 等多种评分,解读门槛稍高 |
| 适用场景 | 快速获取高质量同源模型的首选 | 当 SWISS-MODEL 找不到模板时,作为第一替补 |
小结:如果你有一个明确的同源模板,请毫不犹豫地选择 SWISS-MODEL,因为它更快、更简单。当 SWISS-MODEL 告诉你“找不到模板”时,请转向 I-TASSER。
2. AlphaFold2 (ColabFold) vs SWISS-MODEL
AlphaFold2 是 DeepMind 开发的革命性 AI 工具,彻底改变了结构生物学。ColabFold 是其一个流行的、免费易用的在线版本。
| 对比维度 | SWISS-MODEL | AlphaFold2 (ColabFold) |
|---|---|---|
| 核心算法 | 基于物理和进化的同源建模 | 基于深度学习的端到端预测 |
| 模板需求 | 必须 | 不需要,但可利用模板提升精度 |
| 预测能力 | 仅限有模板区域 | 全蛋白,包括全新折叠和高度柔性区域 |
| 模型精度 | 高(高序列一致性时) | 极高,尤其在无同源模板时,实现革命性突破 |
| 操作门槛 | 极低,网页表单 | 中等,需在 Google Colab 中运行 Notebook |
| 计算资源 | 服务端免费提供 | 依赖 Google 提供的免费 GPU,有使用时长限制 |
| 结果解读 | 直观的 QMEAN 评分 | 需理解 pLDDT 和 PAE 图,对新手有一定难度 |
| 速度 | 快 | 较慢(分钟到小时级) |
| 复合物预测 | 仅同源寡聚体 | 支持蛋白-蛋白、蛋白-核酸等多种复合物 |
小结:AlphaFold2 是更强大、更通用的“瑞士军刀”,在精度和通用性上全面超越。但 SWISS-MODEL 依然凭借其极致的速度、零门槛的操作和直观清晰的结果解读,在日常的、有模板可循的建模任务中占据着不可替代的生态位。它不是 AlphaFold 的竞争对手,而是互补者。事实上,SWISS-MODEL 已经将 AlphaFold DB 作为模板库的一部分,实现了强强联合。
3. 选购决策树
面对众多选择,你可以遵循以下决策路径:
- 你的蛋白在 PDB 中有明显的同源模板吗(例如,BLAST E-value < 1e-10)?
- 是 → 转到 2。
- 否 → 转到 3。
- 你的目标是什么?
- 快速获取一个带质量评估的直观模型,用于指导突变实验或教学。 → 选择 SWISS-MODEL。
- 进行高精度的药物设计,或需要建模蛋白-配体复合物。 → 选择 SWISS-MODEL(获取初步模型),然后结合 AlphaFold2 或等待实验结构。
- 需要批量建模 100 个蛋白。 → 选择 SWISS-MODEL API。
- 你需要预测一个无模板的全新折叠蛋白,或一个复杂的蛋白-核酸复合物吗?
- 是 → 选择 AlphaFold2/3 (ColabFold) 或 RoseTTAFold。
- 否,但 SWISS-MODEL 找不到模板,而你又需要某种结构信息。 → 选择 I-TASSER。
八、 常见问题解答
1. SWISS-MODEL 是免费的吗?
是的,SWISS-MODEL 对全球的学术用户完全免费。你无需支付任何费用即可使用其全部功能,包括自动模式、比对模式、项目模式、API 接口以及访问 SWISS-MODEL 仓库。这项服务由瑞士生物信息学研究所(SIB)和巴塞尔大学资助,旨在促进全球生命科学研究。对于商业用户,则需要联系 SIB 获取商业使用授权。
2. SWISS-MODEL 和 AlphaFold 哪个更好?
这不是一个“谁更好”的问题,而是“哪个更适合你的具体需求”。AlphaFold 在精度和通用性上具有革命性优势,尤其擅长预测无模板的全新结构和复杂复合物。而 SWISS-MODEL 在速度、易用性和结果解读上无出其右。对于一个有明确同源模板的常规蛋白,SWISS-MODEL 能在几分钟内给你一个非常可靠且附带直观质量评估的模型。两者并非替代关系,而是互补关系。最佳实践是:先用 SWISS-MODEL 快速建模,如果结果不满意或没有模板,再求助于 AlphaFold。
3. 如何判断 SWISS-MODEL 生成的模型质量好坏?
SWISS-MODEL 提供了非常直观的质量评估工具。你需要重点关注三个指标:
- 序列一致性:这是最重要的指标。一致性 > 50% 的模型通常非常可靠;30%-50% 的模型,整体折叠可信,但细节需谨慎;< 30% 的模型,仅能作为非常粗略的参考。
- GMQE(全局模型质量估计):一个介于 0 到 1 之间的分数,结合了比对和模板信息,越接近 1 越好。
- QMEAN 评分:这是核心。它会给出一个 Z-score,并与一组高分辨率实验结构进行比较。Z-score 在 -2.0 以上通常是可以接受的。更重要的是,查看局部质量图,它能精确告诉你模型的哪些区域可靠,哪些区域不可靠(例如,长的、无模板支撑的 loop 区通常质量较低)。
九、 结论与下一步行动
在 AI 浪潮席卷结构生物学的 2026 年,SWISS-MODEL 非但没有被淘汰,反而凭借其极致的效率、无与伦比的易用性和深厚的专家系统积淀,牢牢占据着蛋白质同源建模领域不可替代的生态位。它不是一个试图预测万物的黑箱,而是一个透明、可靠、高效的“结构翻译官”,将进化赋予蛋白质的序列保守性,精准地翻译成可供研究的三维结构。
它的核心价值在于速度和确定性。当你有一个需要快速验证的结构假说,当你想为突变实验找到聚焦点,当你需要为虚拟筛选准备一个“够用”的靶点结构时,SWISS-MODEL 就是你最忠诚、最迅捷的伙伴。它完美地填补了从序列到实验结构之间的空白地带,让结构信息不再是少数结构生物学家的特权,而成为所有生命科学研究者都能轻松获取的日常资源。
最终评分:9.3/10
- 易用性:10/10
- 建模速度:9.5/10
- 模型质量(有模板时):9/10
- 功能完整性:8.5/10
- 价格(学术):10/10
- 现代性与扩展性:8/10
现在,你的下一步行动非常明确:
- 打开浏览器,访问官网。
- 找到你手头最感兴趣的那个蛋白的序列或 UniProt ID。
- 点击那个大大的 “Start Modelling” 按钮。
- 在几分钟之内,亲眼见证一条一维的字母序列,如何在你眼前变成一个精妙、美丽的三维分子世界。
当那个三维结构在你眼前旋转的那一刻,你对这个蛋白的理解,将从此不同。





